
The Impact of Comparative Effectiveness Research
President Barak Obama’s *declared* healthcare reform goal was to cut healthcare costs and to provide access to healthcare by mandating healthcare insurance. During the heated debate about *the Affordable Care Act (in 2009, when Obamacare was a pejorative name), two contentious issues in particular aroused public suspicion and these were hotly debated at the time. First, was the proposal for end-of-life consultation panels that were dubbed “death panels” by Senator Charles Grassley. 
Senator Grassley* stated at a town hall meeting: “You have every right to fear. You should not have a government run plan to decide when to pull the plug on grandma. I think that’s a family or religious thing that needs to be dealt with.”[1]
The second contentious issue was so-called “comparative effectiveness research” defined by the Institute of Medicine (IOM, 2009): “Comparative effectiveness research (CER) is the generation and synthesis of evidence that compares the benefits and harms of alternative methods to prevent, diagnose, treat, and monitor a clinical condition or to improve the delivery of care. The purpose of CER is to assist consumers, clinicians, purchasers, and policy makers to make informed decisions that will improve health care at both the individual and population levels”.[2]
Even before comparative effectiveness research was defined by the IOM, it was funded under the American Recovery and Reinvestment Act (ARRA) in March 2009 which provided $1.1 billion for major expansion of CER. CER was sold to congress and other government officials, and to powerful public health institutions, as a cost-saving strategy.[3] Among its prominent promoters who have continued to provide the mantle of academic prestige, is the Harvard School of Public Health which launched “the Dean’s Flagship Initiative in Comparative Effectiveness Research to shape the national dialogue around CER and specifically to broaden its scope to include public health programs and health system interventions.”

Dr. Alan Garber, Dean, Chief Academic Officer, Harvard University
“HSPH [Harvard School of Public Health] considers methods of assessing health value, such as cost-effectiveness analysis and preference-based measures of health improvement such as quality-adjusted life years, to be essential elements of policy-relevant CER. Quantitative, comparative measures of health value will provide decision makers with the best information to make the health care system work more efficiently. Higher-value health care can lead to improved health outcomes while stabilizing or reducing health care expenditures.”[4]
However, the public recognized the potential threat posed by CER to personalized medicine:[5] it would provide the government with the “evidence” to support limiting treatment choices. Suspicion that CER would be used by government to intrude into the patient-physician relationship by setting limits on the freedom to choose therapy in consultation with one’s physician. By setting limits on the freedom to choose a therapy, the government can ration healthcare Rationing, it was reasoned, would be accomplished through government medical reimbursement schedules, such as Medicare, Medicaid, and the Veterans Affairs healthcare.
To counteract these concerns, a massive public relations campaign was spearheaded by the healthcare industry and the Center for American Progress (CAP)– Washington’s most influential “idea factory” during the Obama era –was initiated by John Podesta 
John Podesta, the political consultant and Chief of Staff in the Clinton administration, and Counselor to President Barak Obama. CAP organized more than a dozen health experts to contribute to a 110-page report, which was published in conjunction with the Institute on Medicine as a Profession at Columbia University. Their focus was to promote major changes to the infrastructure and organization of health care delivery, and payment incentives. CAP promoted comparative effectiveness as a better method for evaluating the efficacy of different drugs, medical devices, and clinical procedures — “better health through better information”.[6] [The Center for American Progress (CAP) is opposed to single payer health care.]
Of note, the pharmaceutical industry and the health insurance industry were major lobbyists that lent support to head-to-head comparative effectiveness studies. Indeed, Merck’s senior director for public policy stated that Merck backs comparative effectiveness research:
“We aren’t stepping up a fight with President Obama. Merck continues to encourage Congress to pass a more permanent comparative effectiveness research structure and process. Presenting valid research that people can trust can be a valuable tool for the health-care system.”
- The cost for pharmaceutical industry’s support was the removal of cost-cutting language from the funding provision of the Obama Stimulus package.[7]
Similarly, the health insurance industry supported the Obama CER studies. WellPoint Inc. (the second-largest health insurer by revenue) and Cigna Corp. backed Obama with a lobbying push of their own. The president’s Federal Coordinating Council for Comparative Effectiveness Research included *Dr. Ezekiel Emanuel, his key healthcare adviser who is the brother of Rahm Emanuel, White House Chief of Staff. The president selected Ezekiel to oversee CER studies.

The council’s first report to President Obama and Congress contained recommendations as to what treatments should be studied in CER trials. *Ezekiel, who is a medical doctor, and bioethicist, who was an architect of Obamacare, wrote an essay in The Atlantic (2014) in which he declared that life is not worth living beyond 75 years of age.*
- Healthcare industry lobbyists succeeded in ensuring that nothing in the healthcare legislation addressed price gouging by the healthcare industry.

Soon after Barak Obama was elected president, the Pharmaceutical Research and Manufacturers of America (PhRMA) helped create the Partnership to Improve Patient Care. PhRMA President, Billy Tauzin and Tony Coelho, former U.S. House Democratic leader, and several industry leaders argued against using CER
“as a reason to deny access to specific therapies to specific patients”: “medicines work better for some people on average. You may not be average. Suppose you’re the patient for whom Drug B doesn’t work. The irony is science is taking us in the direction of precision medicine, where we can figure out from [our] genetic code exactly what medicine works for [me].
The Harvard School of Public Health summarizes the major effect CER has had:
“This had a major effect on changing the landscape of government research funding. Government funding for CER has been the catalyst for federal agencies such as the Agency for Healthcare Research and Quality (AHRQ) and the National Institutes of Health (NIH), federal, state and local public health agencies, and even health insurance companies, health care delivery organizations, and pharmaceutical and device manufacturers, to organize themselves to conduct and disseminate comparative effectiveness research.”[8]
Those who have examined the claimed cost-saving features of Obamacare, refer to the claims as “myths”: According to the annual “National Health Expenditures Summary,” published by the Centers for Medicare and Medicaid Services, health care costs have risen between 4.0% and 9.6% a year. In 2009, personal healthcare expenditure (in millions) was $2,114,636; in 2019, that expenditure increased to $3,242,500.
- “From 2018 to 2027, personal healthcare costs are expected to grow at an average annual rate of 5.64%. The personal cost of healthcare in 2019 is projected to hit an all-time high of 3.24 trillion, a 5.1% increase from the projected 2018 costs.”[9]
Randomized Controlled Trials within the Comparative Effectiveness Research Context
Industry-sponsored randomized controlled trials (RCT) are referred to as “the gold standard” by the FDA, industry and industry-supported scientists. RCTs, however, have been the subject of numerous critiques by independent scientists published in major medical journals. The critiques have primarily focused on the preponderance of positive RCT reports, and the failure to report serious adverse effects of the products tested – i.e., drugs, vaccines, and medical devices. By contrast, the serious design flaws of government- sponsored comparative effectiveness RCTs – flaws that have often resulted in excess deaths[10]— have been largely ignored by most “high impact” medical journals – i.e., those that are widely read by practicing physicians.
The protocol design of government-sponsored, CER experiments conducted on critically ill patients, also require randomization. In 2009, NIH David Wendler, PhD, head of Research Ethics at the National Institutes of Health, wrote an article, published in the American Journal of Bioethics, questioning the need to ensure that prospective human subjects of research understand the significance of randomization — even as he acknowledges that randomization is a departure from “the norms of standard medical care“:
Because this process represents a significant departure from the norms of standard medical care, it is widely assumed that potential research participants must understand randomization to give valid informed consent. This assumption, together with data that many research participants do not understand randomization, implies that randomized clinical trials often fail to obtain adequately informed consent.
- For critically ill patients randomization poses serious risk; for these patients randomization without regard for their critical condition poses an increased risk of death. A protocol design whose foreseeable risk is death, is a violation of the Hippocratic ethos and a violation of the ethical principles for research involving human subjects laid down by the Nuremberg Code, and the federal regulations (45 CFR 46).
- One CER experiment that generated a debate that spilled into the public arena was SUPPORT; the NIH-sponsored, deceptively titled *comparative effectiveness experiment.
SUPPORT was conducted in 1,300 premature infants.
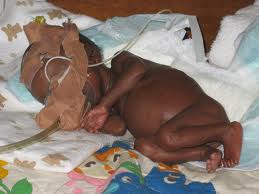
- The mothers were doubly deceived about the nature of the experimental trial; by the deceptive title and the deceptive consent forms which misrepresented the experimental trial as posing no additional risk to the infants, and falsely declaring that the treatment they would receive would be the same as in usual care.[11]Contrary to usual care in infant intensive care units, the infants in the experimental trial were randomized into two extreme levels of oxygen intake.
The flawed design of the experiment and the participation of elite medical institutions; with the approval of every Institutional Review Board (IRB), reveals the scope and magnitude of the problem, namely; the so-called system of human subject protection is a mirage. As Arthur Ammann, MD a clinical professor of pediatrics at the University of California, San Francisco wrote:

“The design and ethical review of federally funded research is often undertaken by a homogeneous group of individuals with congruent interests at the same or similar academic institutions. Individuals from the public, advocacy groups and non-academic rganizations are often excluded. When people from these groups publicly voice their concerns, their views are vilified in academic publications as impeding future advances in research, or even ignored.”
He called for “a far-reaching and comprehensive review of the way US government backed clinical research is funded and approved… The HHS, the NIH and universities must acknowledge that the current research-approval process is flawed and requires an urgent, comprehensive review that should include experts and leaders from outside the academic community.”[12]
In the wake of public condemnation of the conduct of SUPPORT, an NIH-led consortium claimed that “comparative effectiveness research” does not constitute research at all; CER trials, they argued, should be exempt from federal informed consent requirements.[13]
This specious claim was not merely a defensive argument made by those directly involved in the experiment. The argument was endorsed by the pillars of bioethics. Beginning in 2013, these prominent, influential bioethicists argued in a series of articles, that researchers should not be required to obtain explicit consent for research from patients in clinical practice – as is required under those federal regulations – as long as it is known [is vague and undefined] that research is being conducted at the clinic.[14] [15]


Dr.Tom Beauchamp, the principal architect of the Belmont Report (1978) that laid the foundation for the Common Rule – i.e., federal regulations for the protection of human subjects of research. “Traditional informed consent approaches, involving separate discussions and lengthy consent forms, may be an imperfect fit for comparative effectiveness research (CER) that is integrated into usual care and compares non‐investigational treatments.” [16]
Professor Alan Wertheimer, professor emeritus of the University of Vermont, who held visiting appointments at the John F. Kennedy School of Government at Harvard University and Senior Research Scholar at the NIH, argued in the Journal of Law and Biosciences (2014) that “the consent requirement cannot be defended by appeal to any simple principle, such as not treating people merely as a means, bodily integrity, and autonomy…I propose that [sic] that it is legitimate to coerce people into participating in biomedical research.”
He further argued that: “There is some dispute as to whether the ‘reasonable risk’ criterion places any upper limit to the risks to which subjects can be asked to consent, but there is no dispute that institutional review boards (IRBs) must determine that the risks to subjects are reasonable before people are offered the opportunity to consent to participate.”[17]
Although Dr. Wertheimer died in 2015, his perspective about informed consent in the context of medical research is very much alive at the NIH, where the goal of eliminating informed consent requirements from CER trials has been set in motion by two new NIH-sponsored entities that have been joined by the Food and Drug Administration:
NIH- Collaboratory, whose stated mission is: “Strengthen the national capacity to implement cost-effective large-scale research studies that engage healthcare delivery organizations as research partners.”
NIH-Clinical Trials Transformation Initiative (CTTI):
“A public-private partnership whose many stakeholders include government agencies, advocacy groups, professional societies, academic research organizations, and representatives from the medical products industry, CTTI’s mission is to “identify and promote practices that will increase the quality and efficiency of clinical trials.”
On January 18, 2019, the NIH Collaboratory leadership and Demonstration Project Principal Investigators responded to the U.S. FDA’s proposed rule to allow for a waiver or alteration of informed consent.
“We applaud the proposed rule to allow for a waiver or alteration of informed consent for clinical investigations posing no more than minimal risk to a human participant and including appropriate safeguards.
We agree about the broad benefits described in the proposed rule—healthcare advances, reduction in burden from harmonizing FDA’s regulations with the Common Rule, and reduced burden and costs for the IRB…”
- The greatest threat to the integrity of medicine and ethically legitimate medical research is the inordinate pressure exerted by the confluence of powerful interdependent biomedical stakeholders–including academic, commercial and government stakeholders who threaten to weaken, if not eliminate, medical ethics standards.
These ethical standards are embodied in the Hippocratic Oath: “First, do no harm;”
the Nuremberg Code: “The voluntary, informed consent of the human subject is essential”; “No experiment should be conducted where there is an a priori reason to believe that death or disabling injury will occur”;
and the UNESCO Universal Declaration on Bioethics and Human Rights (2005): “Any preventive, diagnostic and therapeutic medical intervention is only to be carried out with the prior, free and informed consent of the person concerned, based on adequate information.”
The Alliance for Human Protection (AHRP) is committed to upholding these inalienable human rights. AHRP was actively involved in calling public attention to ARDS and SUPPORT experiments; we filed letters of complaint to the federal OHRP about the violations of the foremost ethical principle; informed consent. The substance of our complaints was subsequently confirmed by the OHRP investigation. All of the participating medical institutions involved in the ARDS experiment violated the federal requirement for informed consent. Members of the AHRP board also provided testimony about the ethical violations in these trials.
In 2013, AHRP ran a series of articles Medical Research Stakeholders Seek to Overturn Informed Consent Protection:
Part 1: The parameters of ethical research are under pressure
Part 2: Babies in the Crosshairs
Part 3: Bioethicists Promote the Business of Medicine, Not the Ethics of Medicine
References:
[1] Grassley On Death Panels: ‘You Have Every Right To Fear’ By Lisa Lerer, Politico, 2009
[2] The Institute of Medicine (IOM) defined comparative effectiveness research in its report, Initial National Priorities for Comparative Effectiveness (June 2009)
[3] The Role Of Costs In Comparative Effectiveness Research, Alan M. Garber and Harold C. Sox, Health Affairs, 2010]
[4] Comparative Effectiveness Research Initiative, Harvard School of Public Health TH Chan
[5] Alan Garber, MD, PhD, currently Provost and chief academic officer of Harvard University, addressed those fears in an article in The New England Journal of Medicine, titled, “Does Comparative Effectiveness Research Threaten Personalized Medicine?”
[6] Better Health Through Better Information, Ellen-Marie Whelan and Sonia Sekhar, Center for American Progress, 2009
[7] “Makers of drugs and other health-care products say they support side-by-side studies of medical products as a tool for doctors and patients, not as a way to stop medical professionals from prescribing a more expensive drug if they’re convinced it will work better.” Bloomberg News, April 2009
[8] Funding Sources Harvard School of Public Health TH Chan
[9] Average Healthcare Cost in 2019 Student Debt Relief, May 14, 2019; See also, How Has U.S. Spending On Healthcare Changed Over Time? Rabah Kamal and Cunthia Cox, Kaiser Family Foundation, 2019
[10] See: Acute Respiratory Distress Syndrome Network Lower Tidal Volume Trial (ARMA-ARDS): The Case of an Ugly Truth by Rory Spiegel, MD, EMNerd, October 3, 2017; The Surfactant, Positive Pressure, and Pulse Oximetry Randomized Trial (SUPPORT): Another Study Of Preemies Blasted Over Ethical Concerns, Richard Knox, NPR, August 23, 2013; Transfusion Requirements in Critical Care (TRICC): The TRICC Trial, Kent Lavery Reviewer: Chris Nickson, Intensive blog.com 2014; CLOVERS: Report on Research Compliance Volume 16, Number 6. May 22, 2019
[11] Medical Research Stakeholders Seek to Overturn Informed Consent Protections
[12] US Clinical System in Need of Review, Arthur Ammann, NATURE, June 6, 2013
[12b] Ethics and Informed Consent for Comparative Effectiveness Research With Prospective Electronic Clinical Data,Faden, Ruth, PhD, MPH; Kass, Nancy, ScD; Whicher, Danielle, MHS; Stewart, Walter, PhD; Tunis, Sean, MD, MSc Medical Care, 2013
[13] Ethics and Informed Consent for Comparative Effectiveness Research With Prospective Electronic Clinical Data Faden, Ruth, PhD, MPH; Kass, Nancy, ScD; Whicher, Danielle, MHS; Stewart, Walter, PhD; Tunis, Sean, MD, MSc Medical Care, 2013
[14] An Ethics Framework For A Learning Health Care System: A Departure From Traditional Research Ethics And Clinical Ethics, Faden R, Kass NE, Goodman SN, Pronovost P, Tunis S, Beauchamp TL. Hastings Center Reports. 2013; The Research-Treatment Distinction:A Problematic Approach for Determining Which Activities Should Have Ethical Oversight, Nancy E. Kass, Ruth R. Faden, Steven N. Goodman, Peter Pronovost, Sean Tunis and Tom L. Beauchamp, Hastings Center Reports, 2013
[15] Informed Consent For Comparative Effectiveness Trials. Ruth Faden, Tom Beauchamp, Nancy Kass, New England Journal of Medicine, May 2014; Learning Is Not Enough: Earning Institutional Trustworthiness Through Knowledge Translation, Stephanie R. Morain, Nancy E. Kass and Ruth R. Faden, The American Journal of Bioethics, 2018.
[16] Stakeholder Perspectives Regarding Alternate Approaches To Informed Consent For Comparative Effectiveness Research, Stephanie R. Morain, Ellen Tambor, Rachael Moloney, Nancy E. Kass, Sean Tunis, Kristina Hallez, Ruth R. Faden, Learning Health Systems, 2017; Rethinking Informed Consent Requirements for Pragmatic Comparative Effectiveness Trials, Doctoral Dissertation by Danielle Whicher [Johns Hopkins] under guidance of Dr. Nancy Kass and Dr. Ruth Faden; The Ethical Justification For Inclusion Of Neonates In Pragmatic Randomized Clinical Trials For Emergency Newborn Care, DK Kaye (Institute of Bioethics, Johns Hopkins University), BMC Pediatrics, 2019
[17] (Why) Should We Require Consent To Participation In Research? Alan Wertheimer