Who decides if it’s treatment or a medical experiment?
Current worrisome trends
U.S. government and academic bioethicists and institutional ethics review boards (IRB) have expunged the explicit honest terminology of the Nuremberg Code: “experiment” has been replaced by the innocuous term “study”; “human subject” is being replaced by “participant”; and the foremost ethical mandate — “the voluntary, informed consent is absolutely essential”—has been perforated with a series of government waivers from informed consent.
They include: FDA military waivers of consent for testing “investigational new drugs” (1990) has been used to coerce soldiers into accepting the controversial anthrax vaccine; FDA- emergency research waivers (1996) was used to test five experimental artificial blood products in non-consensual trauma patients. All of the blood substitute experiments resulted in causing harm and killing patients.
In 2001, the Department of Health and Human Services amended federal regulation protecting children from research contrary to their best interest by waiving parental permission. Specifically, section 45 CFR 46.408 (c) to provide waivers of parental consent for research involving children.
45 CFR 46.408(c)
“if the IRB determines that a research protocol is designed for conditions or for a subject population for which parental or guardian permission is not a reasonable requirement to protect the subjects (for example, neglected or abused children), it may waive the consent requirements… provided an appropriate mechanism for protecting the children who will participate as subjects in the research is substituted, and provided further that the waiver is not inconsistent with federal, state, or local law.”(Read our Dissenting Opinion)
HHS effectively removed parental judgment and authority — to protect the welfare of their children. A government decree shifted the authority of enrolling children in medical experiments from parents to institutional review boards. These boards’ decisions demonstrate that IRBs protect the institutions that employ them. No IRB has ever assumed responsibility for harm that occurred to a human subject in an IRB-approved clinical trial.
Of note: FDA has not adopted the provisions of 45 CFR 46.408(c) that allow an IRB to waive the requirements for obtaining permission in certain circumstances.
How a medical intervention is defined is meaningful for both patients and physician-scientists. The definition of research carries ethical and legal requirements that researchers must meet.
A heated medical ethics debate in the U.S. was ignited following public disclosure of informed consent violations in major, multi-site, government-sponsored experiments
On one side of the debate are human rights advocates and ethical medical researchers who recognize that the human right laid down by the Nuremberg Code; requiring researchers to obtain “the voluntary, informed consent of the human subject” is “absolutely essential.”

This inviolable human right – the right to decline being a human research subject – is being challenged in the United States by a powerful coalition of research stakeholders whose careers are invested in the business of clinical trials; a business requiring many thousands of human subjects. Following a federal investigation that determined gross violations of informed consent requirements in major NIH sponsored clinical trials, whose foreseeable increased risk was death – ARDS and SUPPORT – high level academic and government officials, led by Dr. Francis Collins, the Director of the National Institutes of Health (NIH), the editor-in-chief of The New England Journal of Medicine; and a posse of academic researchers who are supported by government grants, as well as their entourage of bioethicists are challenging the requirement for informed consent. (Read about the SUPPORT experiment ethics debate) 
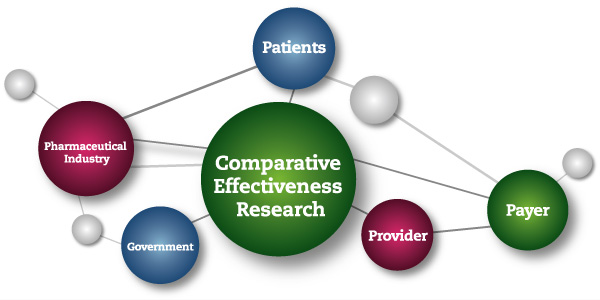
This NIH-led consortium makes the specious claim that controlled clinical trials that they classify as “comparative effectiveness research” (CER) do not constitute research, and should, therefore, be exempt from requirements of informed consent.
The Institute of Medicine of the National Academies of Science, defined CER as follows:
“Comparative effectiveness research (CER) is the generation and synthesis of evidence that compares the benefits and harms of alternative methods to prevent, diagnose, treat, and monitor a clinical condition or to improve the delivery of care. The purpose of CER is to assist consumers, clinicians, purchasers, and policy makers to make informed decisions that will improve health care at both the individual and population levels. ”(Comparative Effectiveness Research,2009)
Comparative Effectiveness Treatment Research is a U.S. government sponsored initiative with an initial $1 billion “stimulus package.” CER trials are being carried out within the framework of research protocols; and CER trial results are reported in medical journals as research findings.